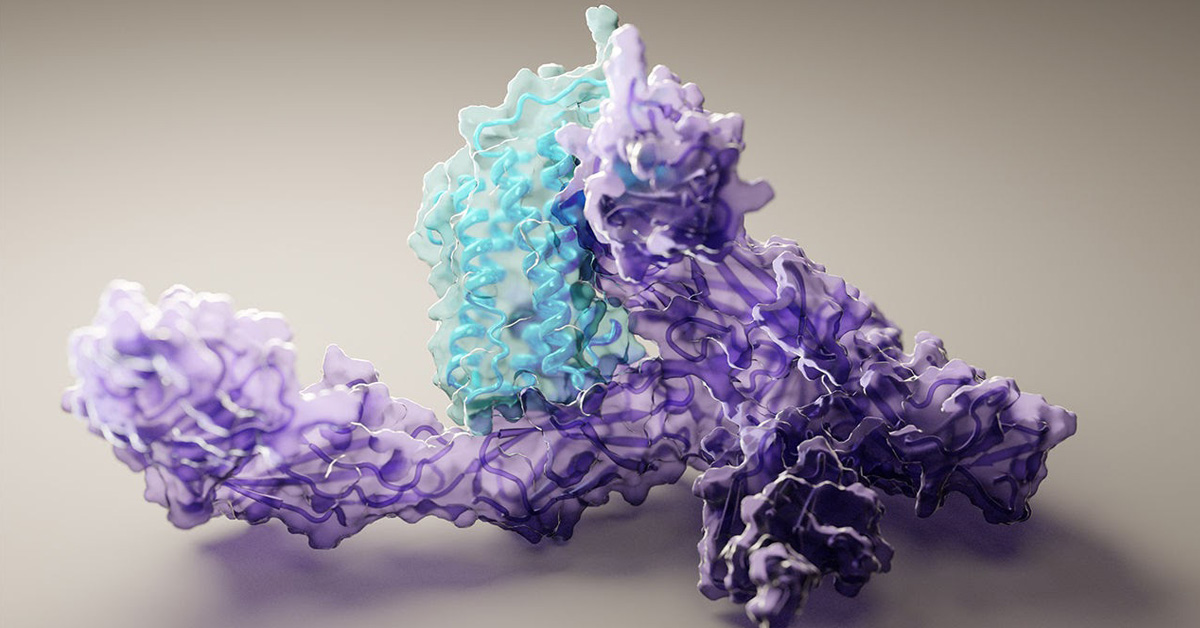
July 15 was a big day for cellular biologists. Software that is highly accurate in predicting protein structures has become broadly accessible to scientists everywhere.
DeepMind, the London-based artificial intelligence (AI) unit of Google that developed the chess master neural network (NN) AlphaZero, released an open-source version of its deep-learning NN AlphaFold 2.0 on GitHub and revealed how they did it in a paper in Nature.
On the same day, an academic team led by scientists from the University of Washington published a paper in Science describing an alternative open-source protein-structure prediction system, called RoseTTaFold, which was also inspired by AlphaFold 2.0. The program, also available on GitHub, was able to achieve similar results as AlphaFold 2.0 and it’s already gaining popularity with scientists.
With the codes open-sourced, the scientific community will be able to freely build on the advances to create even more powerful and useful software, revolutionising the field of cellular biology.
Let’s take a look at how researchers unfold the solution to the protein-folding problem that eluded researchers for over five decades, and understand why it is so important to us.
Deciphering proteins with computers.
Proteins consist of strings of amino acids – the building blocks of everything in our bodies including muscles, hair, antibodies and enzymes. How a protein functions and what it does is determined by how it’s folded into its 3D microscopic shape, underpinning every biological process ever known to us.
‘Structure is function’ is an axiom of cellular biology. Understanding how a protein’s constituent parts – a string of many amino acids – form the many twists and folds of its eventual 3D shape has huge implications. This knowledge allows scientists to accelerate the development of new and better treatments for cancer, pandemics such as COVID-19, and a myriad of other health issues.
For decades, scientists have used many experimental techniques such as X-ray crystallography, which is hailed as the ‘gold standard’ in deciphering protein architecture, and cryo-electron microscopy to unveil the structure of proteins. But these methods are costly and extremely time-consuming.
The rise in big data analytics and AI is helping to fuel progress, allowing computational scientists to apply deep learning algorithms in predicting protein shapes with high accuracy. The modelling software is programmed to harness the ability of AI to distinguish patterns in vast databases of examples, generating ever more informed and accurate iteration as it learns – essentially perfecting itself through self-learning.
To model a new protein, the deep learning approach, run on a couple of gaming GPUs (RTX2080), contrasts the amino acid sequence of the protein to all similar sequences in huge databases. It also concurrently predicts pairwise interactions between amino acids within the protein, while assembling the predicted 3D protein structure at the same time. These steps, also known as ‘tracks’ in the neural network, are bounced back and forth by the AI program to effectively fine-tune the model, using the output of each step to update the others, ultimately resulting in a precise protein structure prediction. More details on the software can be read here.
A whole new window of possibilities.
Both breakthroughs in AlphaFold 2.0 and RoseTTaFold are phenomenal and will undoubtedly usher in a paradigm shift in how scientists use protein structure predictions to advance biology at a more rapid pace. Without the aid of such computational software, it can take years of painstaking laboratory work to ascertain the structure of just one single protein.
DeepMind has entered a partnership with the European Molecular Biology Laboratory (EMBL) to release the most complete and accurate database yet of predicted protein structure models for the human proteome, which will be freely and openly available to the scientific community.
With this game-changing ability to handle multi-protein complexes directly from sequence information, bioinformaticians are now empowered to ask more advanced questions that will herald a new era for AI-enabled biology and give us more insight into the workings of life.